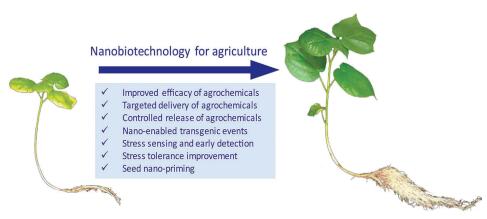
Nano-enabled agriculture is an emerging hot topic. To facilitate the development of nano-enabled agriculture, reviews addressing or discussing the applications, knowledge gap, future research needs, and possible new research field of plant nanobiotechnology in agricultural production are encouraged. Here we review the following topics in plant nanobiotechnology for agriculture: 1) improving stress tolerance, 2) stress sensing and early detection, 3) targeted delivery and controlled release of agrochemicals, 4) transgenic events in non-model crop species, and 5) seed nanopriming. We discuss the knowledge gaps in these topics. Besides the use of nanomaterials for harvesting more electrons to improve photosynthetic performance, they could be used to convert nIR and UV to visible light to expand the light spectrum for photosynthesis. We discuss this approach to maintaining plant photosynthesis under light-insufficient conditions. Our aim in this review is to aid researchers to learn quickly how to use plant nanobiotechnology for improving agricultural production.
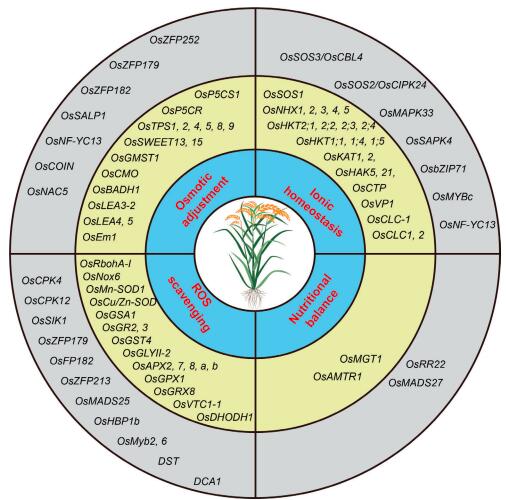
Crop yield loss due to soil salinization is an increasing threat to agriculture worldwide. Salt stress drastically affects the growth, development, and grain productivity of rice (Oryza sativa L.), and the improvement of rice tolerance to salt stress is a desirable approach for meeting increasing food demand. The main contributors to salt toxicity at a global scale are Na+ and Cl- ions, which affect up to 50% of irrigated soils. Plant responses to salt stress occur at the organismic, cellular, and molecular levels and are pleiotropic, involving (1) maintenance of ionic homeostasis, (2) osmotic adjustment, (3) ROS scavenging, and (4) nutritional balance. In this review, we discuss recent research progress on these four aspects of plant physiological response, with particular attention to hormonal and gene expression regulation and salt tolerance signaling pathways in rice. The information summarized here will be useful for accelerating the breeding of salt-tolerant rice.
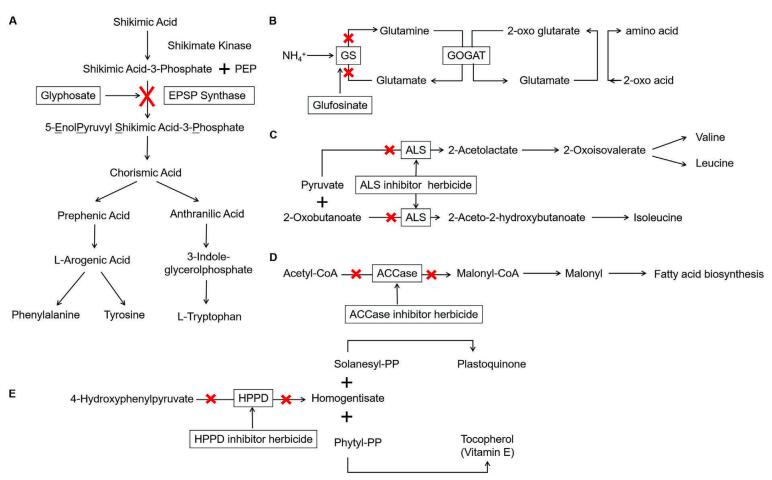
Rice is one of the most important food crops in the world. Weeds seriously affect the rice yield and grain quality. In recent years, there are tremendous progresses in the research and application of herbicide-resistant genes in rice worldwide. This article reviews the working mechanisms of six herbicides (glyphosate, glufosinate, acetolactate synthase inhibitor herbicides, acetyl-CoA carboxylase inhibitor herbicides, hydroxyhenylpyruvate dioxygenase (HPPD) inhibitor herbicides and dinitroaniline herbicides), the resistance mutations of the corresponding herbicide-target genes, and the herbicide detoxification mechanisms by non-target genes. Examples are provided on herbicide-resistant rice materials obtained by transformation of exogenous resistance genes, by artificial mutagenesis and mutant screening, and by modifying the target genes through gene editing. This paper also introduces the current application of herbicide-resistant rice, points out problems that may be caused by utilization of herbicide resistant rice and solutions to the problems, and discusses the future prospects for the development of herbicide-resistant rice.
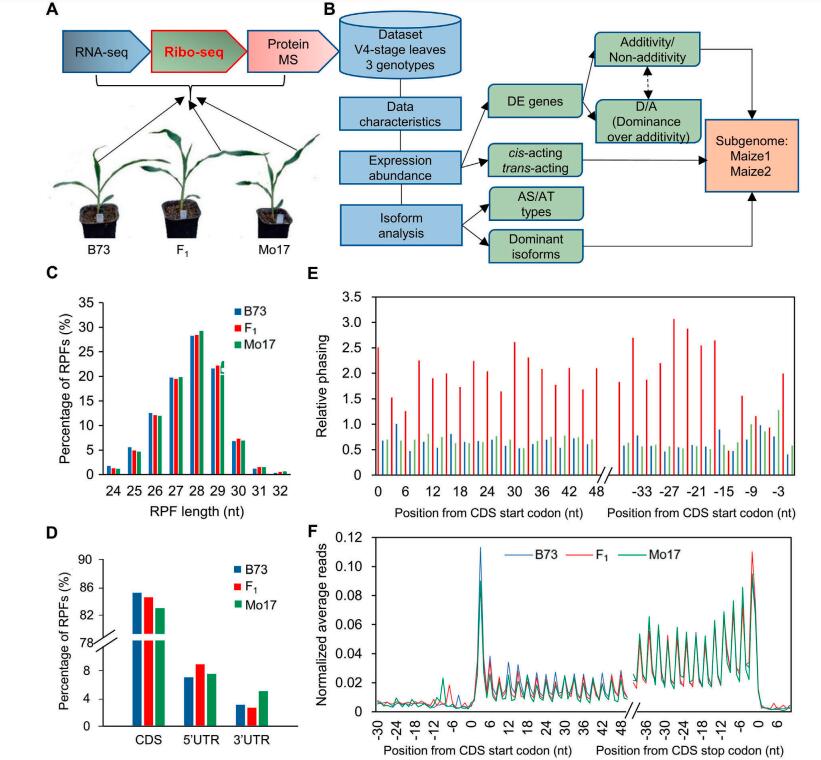
Heterosis, the phenomenon in which hybrids outperform their parents, has been utilized in maize (Zea mays L.) for over 100 years. To provide a more complete understanding of heterosis, we collected a comprehensive transcriptome and translatome dataset on seedling leaves for B73, Mo17, and their F1 hybrid, which provided a dynamic landscape of transcriptomic and translatomic variation in maize. Although additivity accounted for a large proportion of variation at two omics-levels, an elevated nonadditive effect was observed in the translatome, especially in the translated subgenome maize1 genes, and the genes that switched from additivity in the transcriptome to nonadditivity in the translatome were significantly enriched in the subgenome maize1. Many genes with allele-specific expression and translation show dramatic regulatory switches between the transcriptome and translatome, and partial genes with allele-specific translation underlying regulatory mechanism also exhibited subgenome bias. Interestingly, we found the translated isoforms show different expression patterns compared with transcriptome and more genes changed their dominant isoforms during the genetic flow from parents to the hybrid at the translatome level. The translated genes with switched dominant isoforms significantly biased to the subgenome maize2 while genes with conserved dominant isoforms significantly enriched in subgenome maize1. Together, the dynamic changed patterns in translatome across hybrid and parental lines show translational fractionation of the maize subgenomes, which may be associated with heterosis in maize and provides a potential theoretical basis for breeding.
The current assembled maize genomes cannot represent the broad genetic diversity of maize germplasms. Acquiring more genome sequences is critical for constructing a pan-genome and elucidating the linkage between genotype and phenotype in maize. Here we describe the genome sequence and annotation of A188, a maize inbred line with high phenotypic variation relative to other lines, acquired by single-molecule sequencing and optical genome mapping. We assembled a 2210-Mb genome with a scaffold N50 size of 11.61 million bases (Mb), compared to 9.73 Mb for B73 and 10.2 Mb for Mo17. Based on the B73_RefGen_V4 genome, 295 scaffolds (2084.35 Mb, 94.30% of the final genome assembly) were anchored and oriented on ten chromosomes. Comparative analysis revealed that ~30% of the predicted A188 genes showed large structural divergence from B73, Mo17, and W22 genomes, which causes high protein divergence and may lead to phenotypic variation among the four inbred lines. As a line with high embryonic callus (EC) induction capacity, A188 provides a convenient tool for elucidating the molecular mechanism underlying the formation of EC in maize. Combining our new A188 genome with previously reported QTL and RNA sequencing data revealed eight genes with large structural variation and two differentially expressed genes playing potential roles in maize EC induction.

With the increasing promotion of simplified rapeseed cultivation in recent years, the development of cultivars with high resistance to herbicides is urgently needed. We previously developed M342, which shows sulfonylurea herbicide resistance, by targeting acetohydroxyacid synthase (AHAS), a key enzyme in branched-chain amino acid synthesis. In the present study, we used a progeny line derived from M342 for an additional round of ethyl methane sulfonate mutagenesis, yielding the novel mutant DS3, which harbored two mutations in AHAS genes and showed high sulfonylurea resistance. One mutation was the substitution Trp574Leu, as in M342, according to Arabidopsis protein sequencing. The other site was a newly recognized substitution, Pro197Leu. A KASP marker targeting Pro197Leu was developed and reliably predicted the response to sulfonylurea herbicides in the F2 population. The combination of Trp574Leu and Pro197Leu in DS3 produced a synergistic effect that greatly increased herbicide resistance. Analysis of the protein structures of AHAS1 and AHAS3 in wild-type and single-gene mutant plants revealed three-dimensional protein conformational changes that could account for differences in herbicide resistance characteristics including toxicity tolerance, AHAS enzyme activity, and AHAS gene expression.
Manipulation of flowering time to develop cultivars with desired maturity dates is fundamental in plant breeding. It is desirable to generate polyploid rapeseed (Brassica napus L.) germplasm with varying flowering time controlled by a few genes. In the present study, BnaSVP, a rapeseed homolog of the Arabidopsis SVP (Short Vegetative Phase) gene, was characterized and a set of mutants was developed using a CRISPR/Cas9-based gene-editing tool. A single construct targeting multiple sites was successfully applied to precisely mutate four copies of BnaSVP. The induced mutations in these copies were stably transmitted to subsequent generations. Homozygous mutants with loss-of-function alleles and free transgenic elements were generated across the four BnaSVP homologs. All mutant T1 lines tested in two environments (summer and winter growing seasons) showed early-flowering phenotypes. The decrease in flowering time was correlated with the number of mutated BnaSVP alleles. The quadruple mutants showed the shortest flowering time, with a mean decrease of 40.6%-50.7% in length relative to the wild type under the two growth conditions. Our study demonstrates the quantitative involvement of BnaSVP copies in the regulation of flowering time and provides valuable resources for rapeseed breeding.
Cotton architecture is partly determined by shoot branching and flowering patterns. GhBRC1 was previously identified by RNA-seq analysis of nulliplex-branching and normal-branching cotton. However, the roles of GhBRC1 in cotton remain unclear. In the present study, investigations of nuclear localization and transcriptional activity indicated that GhBRC1 has characteristics typical of transcription factors. Gene expression analysis showed that GhBRC1 was highly expressed in axillary buds but displayed different expression patterns between the two branching types. Overexpression of GhBRC1 in Arabidopsis significantly inhibited the number of branches and promoted flowering. In contrast, silencing GhBRC1 in cotton significantly promoted seedling growth. GhBRC1 was induced by multiple hormones, including strigolactones, which promoted seedling growth and seed germination of Arabidopsis plants overexpressing GhBRC1. Consistent with these findings, RNA-seq analysis of virus-induced gene silencing treated cotton revealed that a large number of genes were differentially expressed between GhBRC1-silenced and control plants, and these genes were significantly enriched in plant hormone signalling pathways. Together, our data indicates that GhBRC1 regulates plant branching and flowering through multiple regulatory pathways, especially those regulating plant hormones, with functions partly differing from those of Arabidopsis BRC1. These results provide insights into the molecular mechanisms controlling plant architecture, which is important for breeding cotton with ideal plant architecture and high yield.
Numerous studies using a combination of confocal microscopic- and pharmacological-based approaches have demonstrated that the actin cytoskeleton dynamically responds to pathogen infection. Here, we observed that phalloidin treatment induced actin nucleation, resulting in enhanced resistance of wheat against the stripe rust pathogen Puccinia striiformis f. sp. tritici (Pst). To define the mechanism underpinning this process, we characterized a family of conserved actin-binding proteins, the actin related protein (ARP) family, which controls actin polymerization. Specifically, we identified and characterized a wheat ARPC gene (TaARPC5), which encodes a 136-amino acid protein containing a P16-Arc domain, the smallest subunit of the ARP2/3 complex. TaARPC5 mRNA accumulation was induced following the infection of plants with the avirulent Pst strain, and following the elicitation with flagellin (e.g., flg22) as well. Subcellular localization analysis revealed that TaARPC5 is primarily localized to the cortical actin cytoskeleton, and its precise cellular localizations suggest the proximity to processes correlated with the actin-organelle interface. Upon treatment with virulent Pst, TaARPC5-knockdown plants exhibited a significant reduction in the expression of PTI-specific mRNAs. Conversely, we observed enhanced induction of reactive oxygen species (ROS) accumulation and a decrease in TaCAT1 expression following infection with an incompatible Pst isolate. Together with yeast complementation assays, the current study demonstrates a role for TaARPC5 in resistance signaling in wheat against Pst infection by regulating the host actin cytoskeleton.
Sugar transportation and sugar-to-starch metabolism are considered important processes in seed development and embryo viability. A few plant SWEET proteins acting as sugar transporters have been reported to function in inflorescence and/or seed development. Here, we identified seven members of the 21 OsSWEET genes in rice that play essential roles in sugar transportation and sugar-to-starch conversion in seed development. Nineteen OsSWEET genes exhibiting different expression patterns during inflorescence and seed development were knocked out individually by CRISPR/Cas9. One third of the mutants showed decreased fertile pollen viability and shriveled mature caryopses, resulting in weakened seed traits. Grain fill-related genes but not representative grain shape-regulating genes showed attenuated expression in the mutants. Seed of each of these mutants accumulated more sucrose, glucose or fructose but less starch. Among all OsSWEET genes, OsSWEET4 and OsSWEET11 had major effects on caryopsis development. The sugar-to-starch metabolic pathway was significantly altered in ossweet11 mutants based on differential expression analysis in RNA sequencing assays, confirming that OsSWEET11 functions as a sugar transporter with a key role in seed development. These results help to decipher the multiple functions of OsSWEET genes and to show how they might be used in genetic improvement of rice.
The two most important activities in maize breeding are the development of inbred lines with high values of general combining ability (GCA) and specific combining ability (SCA), and the identification of hybrids with high yield potentials. Genomic selection (GS) is a promising genomic tool to perform selection on the untested breeding material based on the genomic estimated breeding values estimated from the genomic prediction (GP). In this study, GP analyses were carried out to estimate the performance of hybrids, GCA, and SCA for grain yield (GY) in three maize line-by-tester trials, where all the material was phenotyped in 10 to 11 multiple-location trials and genotyped with a mid-density molecular marker platform. Results showed that the prediction abilities for the performance of hybrids ranged from 0.59 to 0.81 across all trials in the model including the additive effect of lines and testers. In the model including both additive and non-additive effects, the prediction abilities for the performance of hybrids were improved and ranged from 0.64 to 0.86 across all trials. The prediction abilities of the GCA for GY were low, ranging between?-?0.14 and 0.13 across all trials in the model including only inbred lines; the prediction abilities of the GCA for GY were improved and ranged from 0.49 to 0.55 across all trials in the model including both inbred lines and testers, while the prediction abilities of the SCA for GY were negative across all trials. The prediction abilities for GY between testers varied from?-?0.66 to 0.82; the performance of hybrids between testers is difficult to predict. GS offers the opportunity to predict the performance of new hybrids and the GCA of new inbred lines based on the molecular marker information, the total breeding cost could be reduced dramatically by phenotyping fewer multiple-location trials.
MicroRNAs (miRNAs) act as regulators of plant development and multiple stress responses. Here we demonstrate that the rice miR171b-SCL6-IIs module regulates the balance between blast resistance, grain yield, and flowering. miR171b-overexpressing rice plants (OX171b) displayed increased rice blast resistance accompanied with enhanced defense responses and late heading, whereas blocking miR171b expression in rice (MIM171) led to greater susceptibility to blast disease, associated with compromised defense responses and early heading. Either overexpressing or silencing of miR171b significantly affected plant height and number of filled seeds per panicle (seed-setting rate), resulting in decreased grain yield. miR171b targets SCL6-IIa, SCL6-IIb, and SCL6-IIc, whose expression was suppressed in OX171b but increased in MIM171. Mutants of SCL6-IIa, SCL6-IIb, and SCL6-IIc all displayed phenotypes like that of OX171b, including markedly increased blast disease resistance, slightly decreased grain yield, and delayed flowering. Amounts of miR171b increased gradually in leaves during the vegetative stage but decreased gradually in panicles during the reproductive stage, whereas SCL6-IIs displayed the reverse expression pattern. Together, these results suggest that the expression of miR171b was time- and space-dependent during the rice growth period and regulated the balance between rice blast disease resistance, grain yield, and flowering via SCL6-IIs, and that appropriate accumulation of miR171b is essential for rice development.
As the main byproduct of cotton production, cottonseed yields edible vegetable oil, ruminant feed, and industrial products. We evaluated the individual and interactive effects of elevated air temperature and soil drought on cottonseed yield and nutritional quality using two cotton cultivars, Sumian 15 (heat-susceptible) and PHY370WR (heat-tolerant). The experiment was conducted under three levels of soil relative water content (SRWC): (75 ± 5)%, (60 ± 5)% and (45 ± 5)% and two temperature regimes: ambient temperature (AT, 31.0/26.4 °C, mean daytime/night temperature) and elevated temperature (ET, 33.4/28.9 °C). Cottonseed yield, boll number, seed number, and single-seed weight were lower under combined ET and SRWC(45 ± 5)% than either individual stress or combined stresses in comparison with the control treatment (SRWC(75 ± 5)% under AT). Drought tended to increase oil content and reduce protein content, whereas ET showed almost the opposite effects. Under the combination of ET and soil drought, oil content was still higher than under control, although ET weakened the beneficial effects of drought. For protein, ET offset the negative impacts of mild drought on protein content, but protein content was not increased under SRWC(45 ± 5)%. Each stress or combined stress reduced oil and protein yields under all treatments, owing to declines in cottonseed yields. The combined stress reduced unsaturated fatty acid (UFA)/saturated fatty acid (SFA) and essential amino acid (EAA)/non-essential amino acid (NAA). Compared with PHY370WR, the sensitivity of Sumian 15 to the combined factors was evidenced in the following ways: (1) seed yield, yield components, oil and protein yields were decreased more for Sumian 15 than PHY370WR compared with the control treatment; (2) the combined stresses caused lower oil content, UFA, and UFA/SFA in Sumian 15 than PHY370WR; (3) interaction effects of the combined factors on protein content and EAA/NAA were detected only in Sumian 15.
Elevated levels of atmospheric CO2 (eCO2) promote rice growth and increase methane (CH4) emissions from rice paddies, because increased input of plant photosynthate to soil stimulates methanogenic archae. However, temporal trends in the effects of eCO2 on rice growth and CH4 emissions are still unclear. To investigate changes in the effects of eCO2 over time, we conducted a two-season pot experiment in a walk-in growth chamber. Positive effects of eCO2 on rice leaf photosynthetic rate, biomass, and grain yield were similar between growing seasons. However, the effects of eCO2 on CH4 emissions decreased over time. Elevated CO2 increased CH4 emissions by 48%-101% in the first growing season, but only by 28%-30% in the second growing season. We also identified the microbial process underlying the acclimation of CH4 emissions to atmospheric CO2 enrichment: eCO2 stimulated the abundance of methanotrophs more strongly in soils that had been previously exposed to eCO2 than in soils that had not been. These results emphasize the need for long-term eCO2 experiments for accurate predictions of terrestrial feedbacks.
Reproductive stage frost poses a major constraint for wheat production in countries such as Australia. However, little progress has been made in identifying key genes to overcome the constraint. In the present study, a severe frost event hit two large-scale field trials consisting of six doubled haploid (DH) wheat populations at reproductive stage (young microspore stage) in Western Australia, leading to the identification of 30 robust frost QTL on 17 chromosomes. The major 18 QTL with the phenotype variation over 9.5% were located on 13 chromosomes including 2A, 2B, 2D, 3A, 4A, 4B, 4D, 5A, 5D, 6D, 7A, 7B and 7D. Most frost QTL were closely linked to the QTL of anthesis, maturity, Zadok stages as well as linked to anthesis related genes. Out of those, six QTL were repetitively detected on the homologous regions on 2B, 4B, 4D, 5A, 5D, 7A in more than two populations. Results showed that the frost damage is associated with alleles of Vrn-A1a, Vrn-D1a, Rht-B1b, Rht-D1b, and the high copy number of Ppd-B1. However, anthesis QTL and anthesis related genes of Vrn-B1a and TaFT3-1B on chromosomes 5B and 1B did not lead to frost damage, indicating that these early-flowering phenotype related genes are compatible with frost tolerance and thus can be utilised in breeding. Our results also indicate that wild-type alleles Rht-B1a and Rht-D1a can be used when breeding for frost-tolerant varieties without delaying flowering time.
Brassinosteroids (BRs) are steroid hormones that function in plant growth and development and response to environmental stresses and nutrient supplies. However, few studies have investigated the effect of BRs in modulating the physiological response to nitrogen (N) supply in maize. In the present study, BR signaling-deficient mutant zmbri1-RNAi lines and exogenous application of 2,4-epibrassinolide (eBL) were used to study the role of BRs in the regulation of physiological response in maize seedlings supplied with N. Exogenous application of eBL increased primary root length and plant biomass, but zmbri1 plants showed shorter primary roots and less plant biomass than wild-type plants under low N (LN) and normal N (NN) conditions. LN induced the expression of the BR signaling-associated genes ZmDWF4, ZmCPD, ZmDET2, and ZmBZR1 and the production of longer primary roots than NN. Knockdown of ZmBRI1 weakened the biological effects of LN-induced primary root elongation. eBL treatment increased N accumulation in shoots and roots of maize seedlings exposed to LN or NN treatment. Correspondingly, zmbri1 plants showed lower N accumulation in shoots and roots than wild-type plants. Along with reduced N accumulation, zmbri1 plants showed lower NO3- fluxes and 15NO3- uptake. The expression of nitrate transporter (NRT) genes (ZmNPF6.4, ZmNPF6.6, ZmNRT2.1, ZmNRT2.2) was lower in zmbri1 than in wild-type roots, but eBL treatments up-regulated the transcript expression of NRT genes. Thus, BRs modulated N physiological response and regulated the transcript expression of NRT genes to promote N uptake in maize.
Oil and protein content and fatty acid composition are quality traits in peanut. Elucidating the genetic mechanisms underlying these traits may help researchers to obtain improved cultivars by molecular breeding. Whole-genome resequencing of a recombinant inbred population of 318 lines was performed to construct a high-density linkage map and identify QTL for peanut quality. The map, containing 4561 bin markers, covered 2032 cM with a mean marker density of 0.45 cM. A total of 110 QTL for oil and protein content, and fatty acid composition were mapped on the 18 peanut chromosomes. The QTL qA05.1 was detected in four environments and showed a major phenotypic effect on the contents of oil, protein, and six fatty acids. The genomic region spanned by qA05.1, corresponding to a physical interval of approximately 1.5 Mb, contains two SNPs polymorphic between the parents that could cause missense mutations. The two SNP sites were employed as KASP markers and validated using lines with extremely high and low oil contents. These sites may be useful in the marker-assisted breeding of peanut cultivars with high oil contents.
Tiller angle (TA) strongly influences plant architecture and grain yield in cereals. However, the genetic basis of TA in wheat is largely unknown. We identified three TA-related quantitative trait loci (QTL). One of them was QTa.sau-2B-769, a major QTL localized on chromosome arm 2BL. QTa.sau-2B-769 was detected in seven environments, explaining 18.1%-51.1% of phenotypic variance. We developed a linked Kompetitive Allele-Specific Polymerase chain reaction (KASP) marker, KASP-AX-108792274, to further validate this locus in three additional populations in multiple environments. QTa.sau-2B-769 increased TA by up to 24.9% in these populations. There were significant and positive correlations between TA and flag leaf angle (FLANG). However, TA was not correlated with plant height or anthesis date, suggesting that expression of QTa.sau-2B-769 is independent of vernalization. TraesCS2B01G583800, a gene known to be involved in leaf angle regulation, was identified as the most likely candidate gene for QTa.sau-2B-769. These results enrich our understanding of the mechanisms regulating wheat TA at maturity and may support precise mapping and cloning of gene(s) underlying QTa.sau-2B-769.
The gluten proteins of wheat grain are responsible for visco-elastic properties of flour, but they also trigger the immune-response of celiac disease. In this work, two low-gliadin RNA interference (RNAi) wheat lines that differ for the promoter driving the silencing (D-hordein and γ-gliadin promoters for D783 and D793 lines, respectively), were characterized at transcriptomic, and protein fraction levels in the grain. Silencing of gliadins provides a readjustment in the grain protein fractions that also affects to the non-gluten proteins (NGP), which were increased in both RNAi lines. Determination of wheat gluten by means of the R5 monoclonal antibody also showed a strong reduction in the content of gluten in both RNAi lines. Moreover, fructans, an oligosaccharide linked with the development of non-celiac wheat sensitivity (NCWS) were also significantly decreased in RNAi lines. The down-regulation of gliadins fractions also impacts to other metabolic processes, particularly on carbohydrate metabolism, enzyme regulator activity and response to stress. Genes and transcription factors regulated by ABA were up-regulated, which could suggest the implication of this phytohormone on the stress response observed in the RNAi lines.
The embryo in maize has a critical role in controlling kernel nutrition components and grain yield. We measured five embryo weight and size traits, six kernel weight and size traits, and five embryo-to-kernel ratio traits in a nested association mapping (NAM) population of 611 recombinant inbred lines (RILs) derived from four inbred lines including the high-oil, giant-embryo line BY815 as the common parent. Using three statistical methods, we identified 5-22 quantitative trait loci (QTL) for each trait, explaining 4.7%-46.7% of the phenotypic variation. The genetic architecture of maize embryo size and its related traits appeared to be dominated by multiple small-effect loci with little epistasis, and the genetic control underlying embryo size appeared to be distinct from that underlying kernel size. A trait-QTL association network included 205 nodes and 439 edges and revealed 28 key loci associated with at least three traits. Cloned maize genes including ZmUrb2, Emp12 and ZmBAM1d, maize orthologs of known rice genes that control seed size including BG1, XIAO and GS9, and 11 maize orthologs of Arabidopsis EMBRYO-DEFECTIVE (EMB) genes were identified as underlying these key loci. Further, the phenotypic and genetic relationships between embryo size and kernel size were evaluated, and genetic patterns for identified loci that control embryo size and its related traits were proposed. Our findings reveal distinct genetic architectures for embryo size, kernel size, and embryo-to-kernel ratio traits and establish a foundation for the improvement of embryo-size-mediated kernel nutrition and grain yield.
The content and composition of wheat storage proteins are the major determinants of dough rheological properties and breadmaking quality and are influenced by cultivation conditions. This study aimed to investigate the effects of water deficit and high N-fertilizer application on wheat storage protein synthesis, gluten secondary structure, and breadmaking quality. Reverse-phase ultrahigh-performance liquid chromatography analysis showed that storage protein and gluten macropolymer accumulation was promoted under both independent applications and a combination of water-deficit and high N-fertilizer treatments. Fourier-transform infrared spectroscopy showed that water deficit and high N-fertilizer treatments generally improved protein secondary structure formation and lipid accumulation, and reduced flour moisture. In particular, high N-fertilizer application increased β-sheet content by 10.4% and the combination of water-deficit and high N-fertilizer treatments increased random coil content by 7.6%. These changes in gluten content and secondary structure led to improved dough rheological properties and breadmaking quality, including superior loaf internal structure, volume, and score. Our results demonstrate that moderately high N-fertilizer application under drought conditions can improve gluten accumulation, gluten secondary structure formation, and baking quality.
The Green Revolution gene sd1 has been used extensively in modern rice breeding, especially in indica cultivars. However, elite sd1 alleles and related germplasm resources used for japonica rice breeding have not been identified, and extensive efforts are needed for japonica rice breeding to obtain new dwarfing sources. Data from MBKbase-Rice revealed seven sd1 haplotypes in indica and four in japonica rice. Two new sd1 alleles were identified in indica rice. In 295 japonica accessions from northeast Asia, except for the weak functional allele SD1-EQ, sd1-r was the major allele, reducing plant height in comparison with SD1-EQ. Japonica germplasm resources carrying reported sd1 alleles were identified by genotype searching and further verified by literature search, genealogical analysis, and dCaps markers. Pedigrees and geographic distribution showed that sd1-r is an excellent allele widely used in northern China and Tohoku in Japan, and sd1-j is commonly used in east China and Kyushu in Japan. Dongnong- and Xiushui-series cultivars carrying sd1-r and sd1-j, respectively, are essential branches of the backbone parents of Chinese japonica rice, Akihikari and Ce21, with the largest number of descendants and derived generations. In semi-dwarf japonica rice breeding, sd1-d was introgressed into Daohuaxiang 2 (DHX2). Dwarf and semi-dwarf lines carrying sd1-d were selected and designated as 1279 and 1280, respectively, after withstanding typhoon-induced strong winds and heavy rains in 2020, and are anticipated to become useful intermediate materials for future genetic research and breeding. This work will facilitate the introduction, parental selection, and marker-assisted breeding, and provide a material basis for the next step in identifying favorable genes that selected together with the sd1 alleles in japonica backbone parents.
Salinity, a major abiotic stress, reduces plant growth and severely limits agricultural productivity. Plants regulate salt uptake via calcineurin B-like proteins (CBLs). Although extensive studies of the functions of CBLs in response to salt stress have been conducted in Arabidopsis, their functions in Setaria italica are still poorly understood. The foxtail millet genome encodes seven CBLs, of which only SiCBL4 was shown to be involved in salt response. Overexpression of SiCBL5 in Arabidopsis thaliana sos3-1 mutant rescued its salt hypersensitivity phenotype, but that of other SiCBLs (SiCBL1, SiCBL2, SiCBL3, SiCBL6, and SiCBL7) did not rescue the salt hypersensitivity of the Atsos3-1 mutant. SiCBL5 harbors an N-myristoylation motif and is located in the plasma membrane. Overexpression of SiCBL5 in foxtail millet increased its salt tolerance, but its knockdown increased salt hypersensitivity. Yeast two-hybrid and firefly luciferase complementation imaging assays showed that SiCBL5 physically interacted with SiCIPK24 in vitro and in vivo. Co-overexpression of SiCBL5, SiCIPK24, and SiSOS1 in yeast conferred a high-salt-tolerance phenotype. Compared to wild-type plants under salt stress conditions, SiCBL5 overexpressors showed lower accumulations of Na+ and stronger Na+ efflux, whereas RNAi-SiCBL5 plants showed higher accumulations of Na+ and weaker Na+ efflux. These results indicate that SiCBL5 confers salt tolerance in foxtail millet by modulating Na+ homeostasis.
Soil inorganic phosphate (Pi) levels are frequently suboptimal for the growth and development of crop plants. Although MADS-box genes participate in diverse plant developmental processes, their involvement in phosphate starvation responses (PSRs) remains unclear. We identified a type I MADS-box transcription factor gene, TaMADS2-3D, which was rapidly induced under low-Pi stress in roots of wheat (Triticum aestivum). A TaMADS2-3D-GFP fusion protein was found located in the nucleus. Transgenic Arabidopsis plants overexpressing TaMADS2-3D (TaMADS2-3DOE) showed shortened primary roots, increased lateral root density, and retarded seedling growth under high-Pi (HP) conditions, accompanied by increased Pi contents in their shoots and roots. The Arabidopsis TaMADS2-3DOE plants showed similar PSR phenotypes under low Pi (LP) conditions. These results indicate constitutive activation of PSRs by overexpression of TaMADS2-3D in Arabidopsis. Reactive oxygen species (ROS), H2O2 and O2-, levels were increased in root tips of Arabidopsis TaMADS2-3DOE plants under HP conditions. Transcriptome analysis of Arabidopsis TaMADS2-3DOE plants under different Pi regimes revealed expression changes for a variety of PSR genes including AtZAT6. Overexpression of TaMADS2-3D in wheat also led to constitutive activation of PSRs. We propose that TaMADS2-3D regulates plant PSRs probably by modulating ROS homeostasis, root development, PSR gene expression, and Pi uptake. This study increases our understanding of plant PSR regulation and provides a valuable gene for improving phosphorus-use efficiency in wheat and other crops.
Maize growth, organ development, and yield formation are highly controlled by the manner in which the plant captures, partition, and remobilizes biomass and phosphorus (P). Better understanding of biomass and P accumulation, partition, and remobilization processes will improve modeling of crop resource use. However, there is still a lack of detailed data to parameterize the modeling of these processes, particularly for modern maize cultivars. A two-year (2016 and 2017) field experiment with three P fertilization treatments (0 (P0), 75 (P75), and 300 (P300) kg P2O5 ha-1) was conducted on a Fluvo-aquic soil (Quzhou, Hebei province, China) to collect data and quantify key processes for a representative modern maize cultivar (Zhengdan 958) widely grown in China. The proportions of biomass and P partitioned into various maize organs were unaffected by P application rate. Zhengdan 958 showed a much lower leaf-senescence rate than older cultivars, resulting in post-silking leaf photosynthesis being sufficient to meet grain biomass demand. In contrast, 50%-85% of leaf P and 15%-50% of stem P accumulated pre-silking were remobilized into grain, in spite of the large proportion of post-silking P uptake. Our results are consistent with the theory that plants use resources according to the priority order of re-allocation from senescence followed by assimilation and uptake, with the re-translocation of reserves last. The results also enabled us to estimate the threshold P concentrations of Zhengdan 958 for modeling crop P demand. The critical leaf P concentration for individual leaves was 0.25%-0.30%, with a corresponding specific leaf P (SLP) of 75-100 mg P m-2. The structural P concentration for leaf was 0.01%, corresponding to an SLP of 3.8 mg P m-2. The maximum P concentrations of leaves and stems were 0.33% and 0.29%. The residual P concentration for stems was 0.006%.
The main defense response to Soybean mosaic virus (SMV) infection in soybean [Glycine max (L.) Merr.] is thought to be blockage of intercellular virus transport by callose deposition on plasmodesmata. But the specific regulatory mechanism remains largely unknown. In this study, we found that hydrogen peroxide (H2O2) signal downstream of NO was associated with the regulation of callose accumulation. Abundant H2O2 was produced on the cell membrane and cell wall in the incompatible combination of soybean cultivar Jidou 7 and SMV strain N3, whereas no obvious H2O2 was observed in the compatible combination of Jidou 7 and strain SC-8. When H2O2 production was inhibited, callose accumulation induced by SMV infection decreased to a level insufficient to restrict virus transport in the incompatible combination. The H2O2-associated transcriptome dynamics of soybean during SMV infection was investigated. Transcriptome and functional analysis using virus-induced gene silencing showed that GmSEOB and GmPAP27, two genes regulated by H2O2, functioned in resistance by positively regulating the accumulation of callose in response to SMV infection. These results lay a foundation for further research on the signal transduction and molecular regulation of callose deposition during soybean resistance to SMV infection.
Soybean (Glycine max [L.] Merr.) is a food and oil crop whose growth and yield are influenced by root and nodule development. In the present study, GmNMHC5 was found to promote the formation of nodules in overexpressing mutants. In contrast, the number of nodules in Gmnmhc5 edited with CRISPR/Cas9 decreased sharply. In 35S:GmNMHC5 mutants, expression levels of genes involved in nodulation were significantly up-regulated. Both in vitro and in vivo biochemical analyses showed that GmNMHC5 directly interacted with GmGAI (a DELLA protein), and the content of gibberellin 3 (GA3) in overexpressing mutants was lower than that in the wild type. These results revealed that GmNMHC5 participates in the classical GA signaling pathway, and may regulate the content of GA3 to match the optimal concentration required for nodule formation, thereby promoting nodulation by directly interacting with GmGAI. A model illustrating the mechanism by which GmNMHC5 promotes soybean nodulation is presented.
Yield loss (YLoss) in the ratoon crop due to crushing damage to left stubble from mechanical harvesting of the main crop is a constraint for wide adoption of mechanized rice ratooning technology. Soil drying before the harvest of the main crop has been proposed to overcome this problem. The objective of this study was to determine the effect of soil drying during the mid-to-late grain filling stage of the main crop on grain yield of the ratoon crop in a mechanized rice ratooning system. Field experiments were conducted to compare YLoss between light (LD) and heavy (HD) soil drying treatments in Hubei province, central China in 2017 and 2018. YLoss was calculated as the percentage of yield reduction in the ratoon crop with the main crop harvested mechanically, relative to the grain yield of the ratoon crop with the main crop harvested manually. In comparison with LD, soil hardness was increased by 42.8%-84.7% in HD at the 5-20 cm soil depth at maturity of the main crop. Soil hardness at 5 and 10 cm depths reached respectively 4.05 and 7.07 kg cm-2 in HD. Soil drying treatment did not significantly affect the grain yield of the main crop. Under mechanical harvesting of the main crop, HD increased the grain yield of the ratoon crop by 9.4% relative to LD. Consequently, YLoss was only 3.4% in HD, in contrast to 16.3% in LD. The differences in grain yield and YLoss between the two soil drying treatments were explained mainly by panicles m-2, which was increased significantly by HD in the track zone of the ratoon crop compared with LD. These results suggest that heavy soil drying practice during the mid-to-late grain filling stage of the main crop is effective for reducing YLoss of the ratoon crop in a mechanized rice ratooning system.